

STEP INTO THE FIRST AXSPA EXHIBIT
Explore the pathobiology of axSpA
The chronic inflammatory pathogenesis of axSpA is complex, multifactorial, and arises from external factors…
…that involve multiple adaptive and innate immune cells and pro-inflammatory cytokines.1-3


LECTURE SERIES WITH PROF. IAIN MCINNES
Get insights on cytokines and advancements in rheumatology
Watch the first video in a lecture series featuring Prof. Iain McInnes, a renowned researcher and clinician in the field of rheumatology, and discover how advancements in molecular medicine have revolutionized our understanding of the pathogenesis of axSpA.
The pathologic processes leading to axSpA
The pathologic processes leading to axSpA are not yet completely understood. However, recent studies have revealed insights into the pathways responsible for initiating and maintaining inflammation leading to various clinical manifestations, and some of the key adaptive and innate immune cells and cytokines involved.1,3-7
The role of cytokines in driving inflammation
The latest evidence shows that several different cytokines, including IL-17A, IL-17F, TNF, IL-12, IL-23, as well as direct or indirect involvement of the JAK/STAT pathway, have distinct roles in driving inflammation in different tissues in SpA. Although the relative contributions of each are complex, important distinctions are emerging about which cytokines contribute to various clinical manifestations and SpA disease phenotypes.3-5,7,8


IL-17 family
The IL-17 cytokine family consists of six members that include IL-17A, IL-17B, IL-17C, IL-17D, IL-17E, and IL-17F.9,13
IL-17A and IL-17F share ~50% structural homology and have a similar pro-inflammatory function, signaling via the same receptor complex.12 IL-17A and IL-17F exist as both homo- and heterodimers, meaning three cytokines exist (IL-17A/A, IL-17A/F, and IL-17F/F), all of which have been shown to contribute to inflammation in axSpA.12,14
There are 5 members of the IL-17 receptor family. Two IL-17 receptors (IL-17RC and IL-17RA) are thought to act in concert to mediate signaling by IL-17A/A, IL-17F/F, and IL-17A/F.11,13
Studies have shown that IL-17A and IL-17F cooperate with other mediators of inflammation, such as TNF, to amplify inflammatory responses.12 The contribution of IL-17A and IL-17F to pathological bone formation has been shown in a human periosteum-derived stem cell model of osteogenic differentiation, indicating the importance of IL-17A and IL-17F in bone pathogenesis.10
Receptor Complex


LECTURE SERIES WITH PROF. IAIN MCINNES
Gain a deeper understanding of the role of the IL-23–IL-17 axis in the pathogenesis of axSpA
Join Prof. Iain McInnes as he delves into the latest science on the importance of the Il-23–Il-17 axis in driving pro-inflammatory cytokine responses in axSpA.
Evidence suggests IL-17 is regulated by IL-23-dependent and IL-23-independent pathways in axSpA
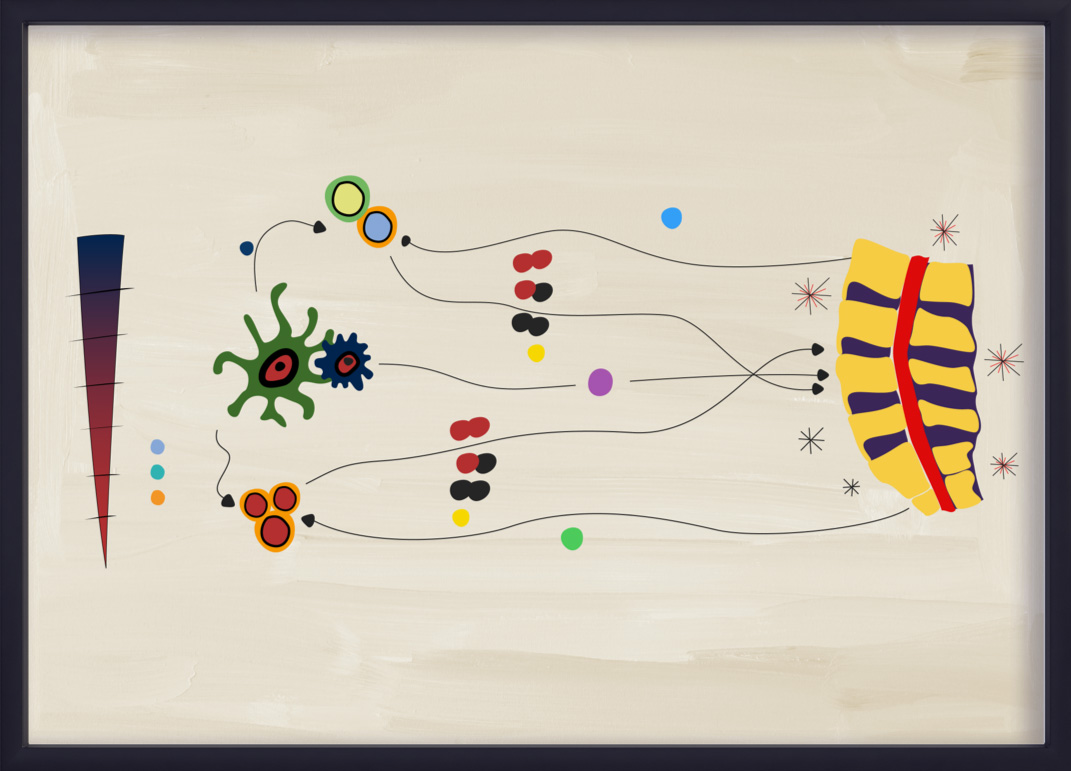
The IL-23–IL-17 axis process
IL-23 is responsible for promoting the expansion and survival of Th17 subsets, including the production of IL-17A and IL-17F from Th17 cells. This process is commonly referred to as the IL-23–IL-17 axis, and has been implicated in several inflammatory diseases, including axSpA.13,14,18,19
Recent evidence suggests that the IL-23–IL-17 axis is not a linear “cascade” in axSpA. IL-23 and IL-17 display partially overlapping but distinct biology and pathobiology.20 Although the IL-23–IL-17 adaptive immunity pathway is still thought to play a role in driving inflammation in axSpA, cells of the innate immune system can also produce IL-17A and IL-17F independently of IL-23.18,20 This aspect of the pathobiology of axSpA may be distinct from PsA, where the IL-23–IL-17 axis still appears to play an integral role in disease.4,19-21
The role of IL-23 in the pathobiology of axSpA is unclear, as clinical trials of biologics blocking this cytokine have failed.
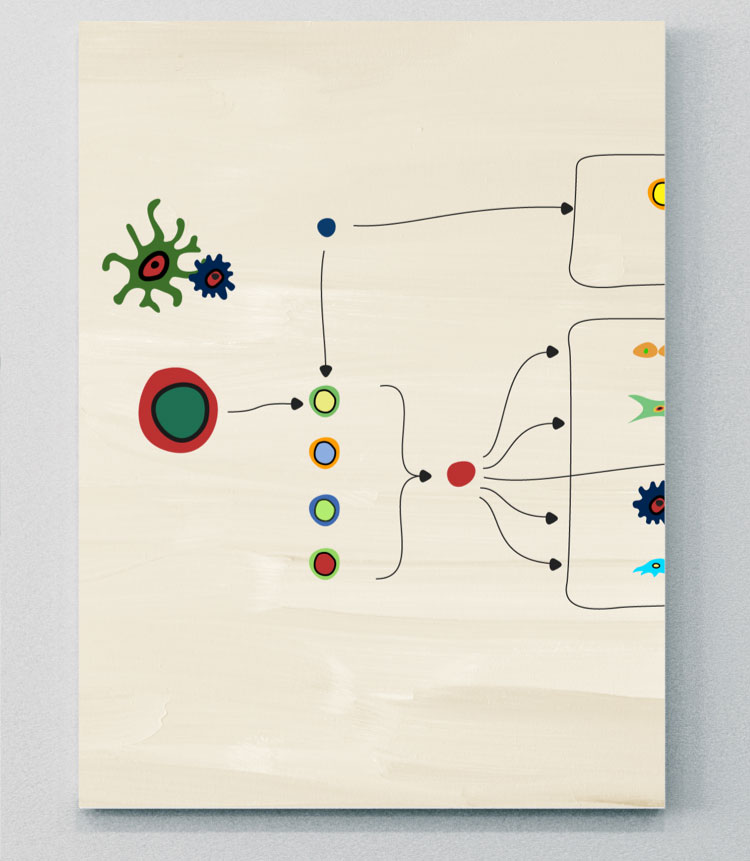

IL-23
An important upstream regulator of IL-1713,20
IL-23 is a member of the IL-12 superfamily and is a heterodimer consisting of both a p40 and p19 chain.13,20 It is an important cytokine involved in the pathogenesis of several immune-mediated inflammatory diseases, including psoriasis, PsA, axSpA, and IBD.20 However, its interaction with different cells and downstream cytokine pathways may be different across diseases.20
Some evidence shows that IL-23 can migrate from barrier sites such as the gut and skin, where dendritic cells have been activated, and travel to sites of disease pathogenesis in axSpA, although this needs to be further explored.20 A key pro-inflammatory role of IL-23 is stimulation of Th17 cells to produce IL-17, IL-22, and TNF, which leads to inflammation, bone formation, and bone erosion.4,13,20
Historically, the IL-23–IL-17 axis has been deemed a central component driving pathological bone formation in SpA diseases, but as evidence evolves, it’s becoming clear that the pathological processes leading to bone loss or formation may vary at different bone sites and across different disease types.4,20


IL-12
Role of IL-12 in axSpA
IL-12 is a pro-inflammatory cytokine composed of two subunits (p35 and p40), resulting in an active heterodimer p70.24 Produced by innate immune cells, it induces naïve T cell differentiation into IFN-gamma producing Th1 cells and prevents T cell exhaustion.24,25
IL-12 and IL-23 activity are tightly linked. Both of these inflammatory cytokines share the p40 subunit and a receptor chain (IL-12R1), which controls distinct IL-12 and IL-23 signaling pathways.24
In axSpA, over-activation of IL-12 and IL-23 increases the number of circulating Th1 and Th17 cells. This activates signaling from cytokines and proteins, which promotes monocyte differentiation into osteoclasts and contributes to sacroiliac and peripheral joint damage.1,26,27
The role of IL-12 in the pathobiology of axSpA is unclear, as axSpA clinical trials of biologics blocking this cytokine have failed.


TNF
TNF signaling plays a role in the pathogenic processes in axSpA
The TNF superfamily (TNFSF) consists of 19 structurally related pleotropic cytokines.29,30 TNFSF proteins are key drivers of inflammation and play roles in the mediation of apoptosis, angiogenesis, cell proliferation, and other critical biologic functions.29
TNF-α exists in transmembrane and soluble forms and is mostly created by T-cells, NK cells, and macrophages.31 TNF-α cellular signaling occurs through two receptors—TNF-receptor1 (TNFR1) and TNFR2—which have differing mechanisms of cellular signaling, ligand affinity, and patterns of expression.32,33 TNFR1 is the dominant receptor involved in inflammatory and innate immune responses.28
TNF-mediated pro-inflammatory TNFR1 signals can increase catabolic bone resorption in the spine by means of osteoclast formation and matrix metalloproteinase production, causing bone erosion.28 Furthermore, TNFR1 and TNFR2 signaling (with other cytokine involvement) can lead to pathogenic bone formation indicative of spinal fusion through increased osteoblast activity.28
In axSpA, TNF-mediated signaling plays an important role in disease progression.
Activated dendritic cells can secrete TNF-α, which leads to increased T-cell signaling and Th cell differentiation.30 Elevated levels of TNF can be found in the synovium and other locations on the body such as the sera and aqueous humor of patients with anterior uveitis.34
In axSpA, TNF can synergize with IL-17 and IL-23 pathways.
- IL-23 stimulates Th17 cell differentiation, which leads to the production of cytokines such as IL-17 and causes further upregulation of TNF.1
- IL-17 can combine with TNF, leading to stronger pro-inflammatory cytokine expression (e.g., IL-6 and IL-8) than either cytokine can enact alone.35
TNF=tumor necrosis factor.


JAK-STAT
JAK-STAT pathways may play an important role in the pathogenesis of axSpA
JAK molecules are a group of intracellular tyrosine kinases, comprising four isoforms: JAK1, JAK2, JAK3, and tyrosine kinase (TYK) 2.37 These groups are coupled to signal transducer and activator of transcription (STAT) molecules.5 Many immune cells and effector molecules use different combinations of JAK and STAT molecules to convert signals from the cell surface to the nucleus, where they activate transcription and induce gene activation. This downstream gene activation has a role in regulating different biological processes, including the activation of pathological pathways in axSpA.5,36,38 Many SpA-associated cytokines are mediated either directly or indirectly via JAK-STAT pathways.36
JAK=Janus kinase.

Discover how several different cytokines have distinct roles in driving inflammation in PsA


NEXT ROOM IN THE PATHOBIOLOGY EXHIBIT
The Pathobiology Learning Theatre
Immerse yourself in the science of IL-17A and IL-17F and their role in the inhibition of inflammation in axSpA.
Become a member of the RheuMuseum and be the first to know about new exhibits
- Furst DE, Louie JS. Targeting inflammatory pathways in axial spondyloarthritis. Arthritis Res Ther. 2019;21(1):135. Published 2019. doi:10.1186/s13075-019-1885-z
- Watad A, Bridgewood C, Russell T, et al. The early phases of ankylosing spondylitis: emerging insights from clinical and basic science. Front Immunol. 2018;9:2668. doi: 10.3389/fimmu.2018.02668
- Rezaiemanesh A, Abdolmaleki M, Abdolmohammadi K, et al. Immune cells involved in the pathogenesis of ankylosing spondylitis. Biomed Pharmacother. 2018;100:198-204. doi:10.1016/j.biopha.2018.01.108
- McGonagle DG, McInnes IB, Kirkham BW, et al. The role of IL-17A in axial spondyloarthritis and psoriatic arthritis: recent advances and controversies. Ann Rheum Dis. 2019;78:1167–1178
- Hammitzsch A, Lorenz G, Moog P. Impact of janus kinase inhibition on the treatment of axial spondyloarthropathies. Front Immunol. 2020;11:591176. Published 2020. doi:10.3389/fimmu.2020.591176
- Schwartzman S, Ruderman EM. A road map of the axial spondyloarthritis continuum. Mayo Clin Proc. 2022 Jan;97(1):134-145. doi: 10.1016/j.mayocp.2021.08.007.
- Rosine N, Miceli-Richard C. Innate cells: the alternative source of IL-17 in axial and peripheral spondyloarthritis?. Front Immunol. 2021;11:553742. Published 2021. doi:10.3389/fimmu.2020.553742
- Siebert S, Millar NL, McInnes IB. Why did IL-23p19 inhibition fail in AS: a tale of tissues, trials or translation? Ann Rheum Dis. 2019;78(8):1015-1018. doi:10.1136/annrheumdis-2018-213654
- Yang XO, Chang SH, Park H, et al. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008;205(5):1063-1075. doi:10.1084/jem.20071978
- Shah M, Maroof A, Gikas P, et al. Dual neutralisation of IL-17F and IL-17A with bimekizumab blocks inflammation-driven osteogenic differentiation of human periosteal cells. RMD Open. 2020;6(2):e001306. doi:10.1136/rmdopen-2020-001306
- Goepfert A, Lehmann S, Blank J, et al. Structural analysis reveals that the cytokine IL-17F forms a homodimeric complex with receptor IL-17RC to drive IL-17RA-independent signaling. Immunity. 2020;52(3):499-512.e5. doi:10.1016/j.immuni.2020.02.004
- Glatt S, Baeten D, Baker T, et al. Dual IL-17A and IL-17F neutralisation by bimekizumab in psoriatic arthritis: evidence from preclinical experiments and a randomised placebo-controlled clinical trial that IL-17F contributes to human chronic tissue inflammation. Ann Rheum Dis. 2018;77(4):523-532. doi:10.1136/annrheumdis-2017-212127
- Tsukazaki H, Kaito T. The role of the IL-23/IL-17 pathway in the pathogenesis of spondyloarthritis. Int J Mol Sci. 2020;21(17):6401. Published 2020. doi:10.3390/ijms21176401
- Bridgewood C, Sharif K, Sherlock J, et al. Interleukin-23 pathway at the enthesis: The emerging story of enthesitis in spondyloarthropathy. Immunol Rev. 2020;294(1):27-47. doi:10.1111/imr.12840
- Blanco P, Palucka AK, Pascual V, Banchereau J. Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytokine Growth Factor Rev. 2008;19(1):41-52. doi:10.1016/j.cytogfr.2007.10.004
- Cole S, Murray J, Simpson C, et al. Interleukin (IL)-12 and IL-18 synergize to promote MAIT Cell IL-17A and IL-17F production independently of IL-23 signaling. Front Immunol. 2020;11:585134. Published 2020. doi:10.3389/fimmu.2020.585134
- Russell T, Watad A, Bridgewood C, et al. IL-17A and TNF modulate normal human spinal entheseal bone and soft tissue mesenchymal stem cell osteogenesis, adipogenesis, and stromal function. Cells. 2021;10(2):341. Published 2021. doi:10.3390/cells10020341
- McGonagle D, Watad A, Sharif K, et al. Why inhibition of IL-23 lacked efficacy in ankylosing spondylitis. Front Immunol. 2021;12:614255. Published 2021. doi:10.3389/fimmu.2021.614255
- Taams LS, Steel KJA, Srenathan U, et al. IL-17 in the immunopathogenesis of spondyloarthritis. Nat Rev Rheum. 2018;14(8):453–466. Published 2018. doi: 10.1038/s41584-018-0044-2
- Mease P, Van den Bosch F. IL-23 and axial disease: do they come together? Rheumatology (Oxford). 2021;60(Suppl 4):iv28-iv33. doi:10.1093/rheumatology/keab617
- Baeten D, Adamopoulos IE. IL-23 Inhibition in ankylosing spondylitis: where did it go wrong?. Front Immunol. 2021;11:623874. Published 2021. doi:10.3389/fimmu.2020.623874
- Boutet MA, Nerviani A, Gallo Afflitto G, et al. Role of the il-23/il-17 axis in psoriasis and psoriatic arthritis: the clinical importance of its divergence in skin and joints. Int J Mol Sci. 2018;19(2):530. Published 2018. doi:10.3390/ijms19020530
- Zhu W, He X, Cheng K, et al. Ankylosing spondylitis: etiology, pathogenesis, and treatments. Bone Res. 2019;7:22. doi: 10.1038/s41413-019-0057-8.
- Schurich A, Raine C, Morris V, et al. The role of IL-12/23 in T cell-related chronic inflammation: implications of immunodeficiency and therapeutic blockade. Rheumatology (Oxford). 2018;57(2):246-254. doi:10.1093/rheumatology/kex186
- Trinchieri G. Interleukin-12: a pro-inflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu Rev Immunol. 1995;13:251-76. doi: 10.1146/annurev.iy.13.040195.001343
- Poddubnyy D, Jadon DR, Van den Bosch F, et al. Axial involvement in psoriatic arthritis: An update for rheumatologists. Semin Arthritis Rheum. 2021;51(4):880-887. doi:10.1016/j.semarthrit.2021.06.006
- Chyuan IT, Lai JH. New insights into the IL-12 and IL-23: From a molecular basis to clinical application in immune-mediated inflammation and cancers. Biochem Pharmacol. 2020;175:113928. doi:10.1016/j.bcp.2020.113928
- Lata M, Hettinghouse AS, Liu CJ. Targeting tumor necrosis factor receptors in ankylosing spondylitis. Ann N Y Acad Sci. 2019;1442(1):5-16. doi: 10.1111/nyas.13933
- Aggarwal BB, Gupta SC, Kim JH. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood. 2012;119(3):651-665. doi:10.1182/blood-2011-04-325225
- Croft M, Siegel RM. Beyond TNF: TNF superfamily cytokines as targets for the treatment of rheumatic diseases. Nat Rev Rheumatol. 2017;13(4):217-233. doi:10.1038/nrrheum.2017.22
- Jang DI, Lee AH, Shin HY, et al. The role of tumor necrosis factor alpha (TNF-α) in autoimmune disease and current TNF-α inhibitors in therapeutics. Int J Mol Sci. 2021;22;2719. https://doi.org/10.3390/ijms22052719
- Holbrook J, Lara-Reyna S, Jarosz-Griffiths H, et al. Tumour necrosis factor signaling in health and disease. F1000Res. 2019; 8:F1000 Faculty Rev-111. doi: 10.12688/f1000research.17023.1
- Mantravadi S, Ogdie A, Kraft WK. Tumor necrosis factor inhibitors in psoriatic arthritis. Expert Rev Clin Pharmacol. 2017;10(8):899-910. doi:10.1080/17512433.2017.1329009
- Ebrahimiadib N, Berijani S, Ghahari M, et al. Ankylosing spondylitis. J Ophthalmic Vis Res. 2021;16(3):462-469. doi: 10.18502/jovr.v16i3.9440
- Noack M, Beringer A, Miossec P. Additive or synergistic interactions between IL-17A or IL-17F and TNF or IL-1β depend on the cell type. Front Immunol. 2019;10:1726. Published 2019. doi:10.3389/fimmu.2019.01726
- McInnes IB, Szekanecz Z, McGonagle D, et al. A review of JAK-STAT signaling in the pathogenesis of spondyloarthritis and the role of JAK inhibition [published online ahead of print, 2021 Oct 20]. Rheumatology (Oxford). 2021;keab740. doi:10.1093/rheumatology/keab740
- White JPE, Coates LC. JAK1 selective inhibitors for the treatment of spondyloarthropathies. Rheumatology (Oxford). 2021;60(Suppl 2): ii39-ii44. doi:10.1093/rheumatology/keaa815
- Fragoulis GE, McInnes IB, Siebert S. JAK-inhibitors. New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology (Oxford). 2019;58(Suppl 1):i43-i54. Published 2019. doi: 10.1093/rheumatology/key276